SARS-COV-2: HIGHER EFFICIENCY IN HUNTING VARIANTS
A new mathematical method enables faster detection of viral lineages, maintaining high precision, has been developed to contribute to the worldwide effort aiming at tracing SARS-CoV-2 and deciphering its evolution
The current fight against the pandemic requires to understand evolutionary relationships in order to identify new variants and evaluate their diffusion. Accuracy and speed of genomic investigations are fundamental elements of the surveillance activities performed in the Istituto Zooprofilattico Sperimentale dell’Abruzzo e del Molise (IZSAM).
Researchers from IZSAM’s " National Reference Center for Genomic Sequences of Pathogenic Microorganisms: Database and Bioinformatics Analysis " (GENPAT) developed a new mathematical method to analyse and compare the genomes of large amount of viral samples. Published in the scientific journal BMC Genomics, the proposed algorithm enables researchers to identify viral strains and to track their evolution (so-called phylogenomic analysis) with a comparable precision and faster than the methods already used.
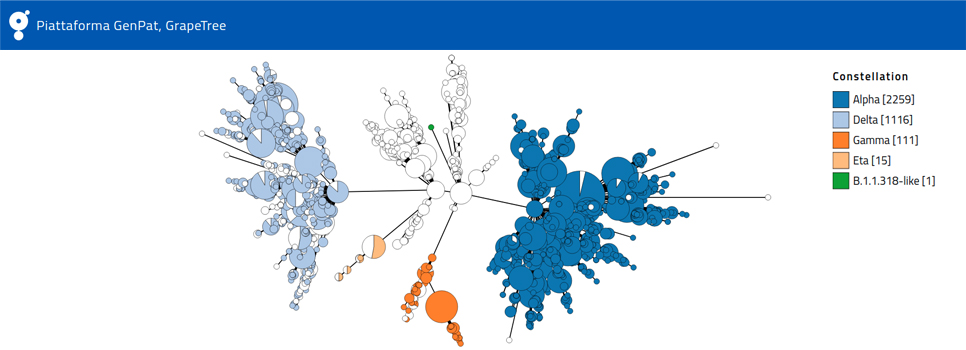
The hunt for variants is a very complex process, starting with simple swabs. Genetic code of the collected viral samples is sequenced with high throughput sequencing technologies (NGS, Next Generation Sequencing) in order to make comparisons between viruses. The aim is to find similarities or differences and, for the latter, to establish genetic "distances" between one strain and another. The result of this mathematical process, allows classification of viral variants according to the international nomenclature "PANGO". Genetic distances between viral genomes can also be graphically represented through tree branches telling us the evolution story of viral lineages, and , to some extent, how it is spreading.
“The procedures commonly used to identify viral lineages- says Adriano Di Pasquale, GENPAT National Reference Center – are fast, an important advantage when you are dealing with a pandemic, but they are not very precise. In other words, they give us a portrait of variants and their evolution, but it is not very detailed. Increasing accuracy through phylogenomic analysis, on the other hand, takes a very long time, even using powerful computers".
The algorithm developed by IZSAM researchers significantly increases the resolution of genomic investigations while maintaining the necessary speed. “In addition to decipher the diffusion of variants - adds Nicolas Radomski, GENPAT - we can now follow their transmission, both in terms of time and space, observing step by step the developing branches”.
Based on the analysis of viral samples collected in Abruzzo and analysed by IZSAM, the new mathematical procedure is now being proposed internationally. “We think it can be a valid tool for all the institutions operating in the context of the pandemic. - concludes Di Pasquale - This is why we already made it available to everyone, on the medRxiv portal. We found considerable interest from various institutions, and a collaboration with the Portuguese National Institute of Health (INSA) is already underway".

- Adriano Di Pasquale
Di Pasquale, A., Radomski, N., Mangone, I., Calistri, P., Lorusso, A., & Cammà, C. 2021. SARS-CoV-2 surveillance in Italy through phylogenomic inferences based on Hamming distances derived from pan-SNPs,-MNPs and-InDels. BMC genomics, 22(1).
")
")
")
")
Istituto Zooprofilattico Sperimentale
dell'Abruzzo e del Molise "G. Caporale"
Campo Boario | 64100 TERAMO | ITALIA
Telefono 0039.0861.3321 | Fax 0039.0861.332251
e-mail: archivioeprotocollo@izs.it
Posta elettronica certificata: protocollo@pec.izs.it
Partita IVA: 00060330677
Codice Fiscale: 80006470670

")